项目展示
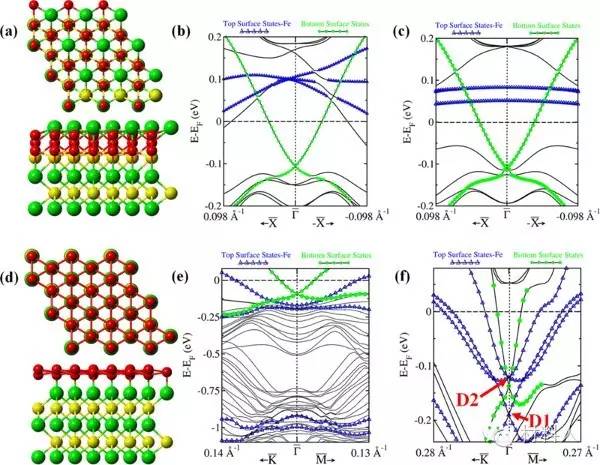
量子点电子结构模拟
使用密度泛函理论计算研究量子点的电子结构和光学性质,为新型光电器件设计提供理论支持。
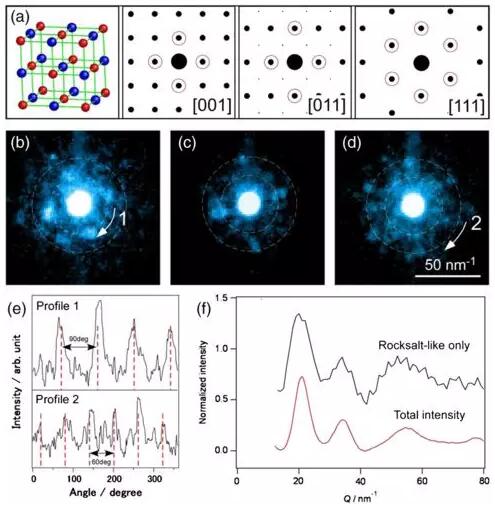
多主元合金相稳定性预测
结合第一性原理计算和机器学习方法,开发多主元合金相稳定性预测模型,加速新型高性能材料设计。
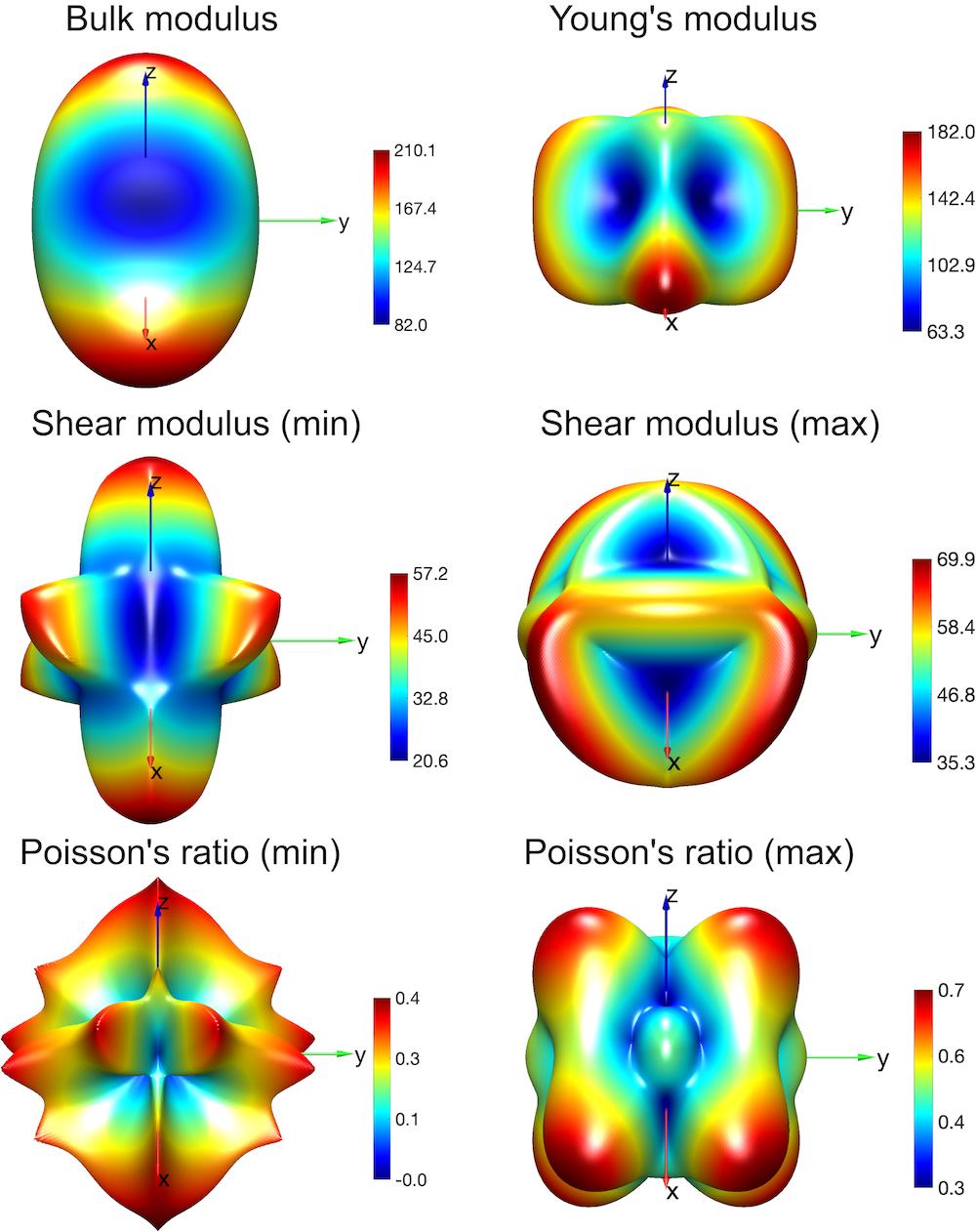
材料力学性能可视化
开发材料力学性能三维可视化工具,直观展示弹性常数、杨氏模量等物理量的空间分布特征。
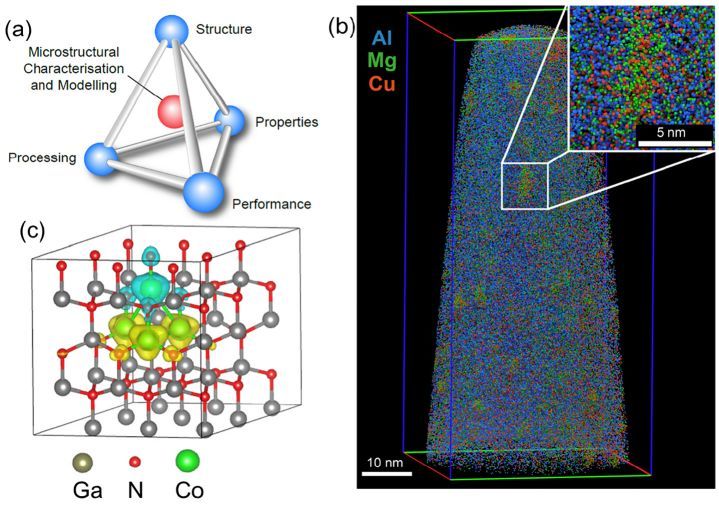
纳米复合材料界面设计
通过分子动力学模拟研究纳米复合材料的界面结构和性能,设计具有优异力学和热学性能的新型复合材料。

量子点显示材料优化
研究量子点材料的发光机制,优化其光学性能,为下一代高清晰度、高色彩饱和度的显示技术提供材料基础。

二维材料电子器件模拟
使用第一性原理计算和量子输运模拟,研究二维材料在纳米电子器件中的应用潜力,设计高性能场效应晶体管。